Batten disease is the common name for a broad class of rare, fatal, inherited disorders of the nervous system also known as neuronal ceroid lipofuscinoses, or NCLs.
- It is an inherited metabolic disorder.
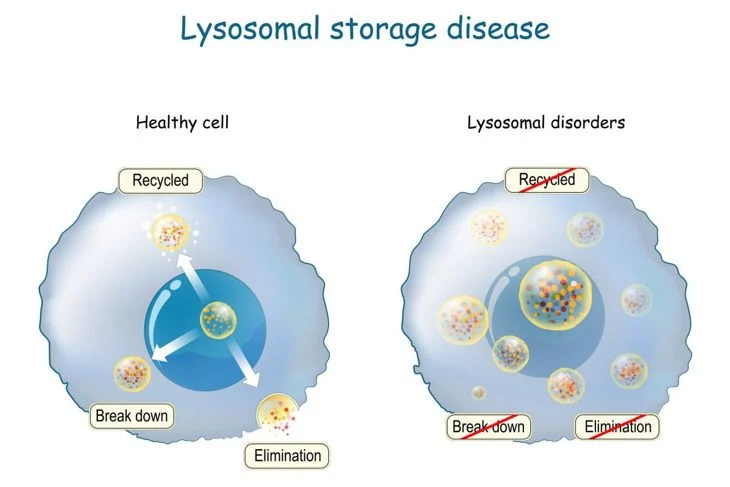
- It is a defect in a specific gene triggers a cascade of problems that interferes with a cell’s ability to recycle certain molecules (lysosomes).
- Lysosomes are the “recycle bin” of the cell and regularly break down waste, proteins, sugars, and naturally occurring fatty compounds called lipids (fatty acids, oils, waxes, and sterols) into smaller components that can be discarded out of the cell or recycled.
- In Batten disease/NCLs, the mutated genes do not produce the proper amounts of proteins important for lysosomal function.
- Each gene (representing a form of the disease) provides information for a specific protein that is in turn, defective and not produced.
- These proteins are needed for brain cells (neurons) and other cells to work efficiently.
- The lack of a functional protein causes the abnormal buildup of “junk (protein and lipids)” material in the lysosomes—as well as the abnormal buildup of the residue called lipofuscin that occurs naturally as part of the lysosomal breakdown of lipids
- The disease has several forms that share some of the same features and symptoms but vary in severity and age when symptoms first begin to appear.
- Each form is caused by a mutation in a different gene.
- Most forms of Batten disease/NCLs usually begin during childhood. Children with the disease often appear healthy and develop normally before they begin to show symptoms.
- Symptoms can appear before the age of one years old.
- Very rarely, adults can develop symptoms (usually around age 30).
- This buildup causes problems with the nervous system that eventually leads to death.
Life expectancy:
- Children with infantile Batten disease die prematurely, often in early childhood.
- Later-onset forms may live into their teens to their thirties.
- If the disease develops in adulthood, the symptoms tend to be milder and may not affect life expectancy.
There are 13 types of neuronal ceroid lipofuscinoses (NCL) and each type depends on what gene is affected.
- Each gene affect different protein function and hence some share similar symptoms and some may appear unique to that type.
Examples include:
CLN1 disease, infantile onset
- The CLN1 gene, found on chromosome 1, directs the production of an enzyme called palmitoyl-protein thioesterase 1 (PPT1). (A chromosome is a threadlike structure that contains all the genetic information needed and resides inside the nucleus of most cells).
- A deficiency in the PPT1 protein or its poor operation allows the abnormal buildup of lipids and proteins.
- In the classic infantile form, symptoms are seen before age 1 and progress rapidly.
- Developmental skills such as standing, walking, and talking are not achieved or are gradually lost.
- Children often develop seizures by age 2 and eventually become blind.
- By age 3 children may become completely dependent on their caregivers, and some may need a feeding tube.
- Most affected children die in early to mid-childhood.
CLN1 disease, juvenile onset
- Some children with CLN1 abnormalities develop the disease after infancy—around age 5 or 6—and have slower disease progression.
- Affected children may live into their teenage years.
- Others may not develop symptoms until adolescence and may live into adulthood.
CLN2 disease, late-infantile onset
- The CLN2 gene, found on chromosome 11, produces an enzyme called tripeptidyl peptidase 1 that breaks down proteins.
- The enzyme is insufficiently active in CLN2 disease.
- Developmental delay begins around the end of age 2.
- Children develop seizures and begin to gradually lose the ability to walk and speak.
- Brief, involuntary jerks in a muscle or muscle group (called myoclonic jerks) typically begin around age 4-5.
- By age 6 most children are completely dependent on their caregivers, and many will require a feeding tube.
- Most children with CLN2 disease die between the ages of 6–12 years.
CLN2 disease, later-onset
- Some children with CLN2 abnormalities develop the disease later in childhood —around age 6 or 7—and have slower disease progression.
- In later-onset CLN2 disease, loss of coordination (ataxia) may be the initial symptom. Affected children may live into their teenage years.
CLN3 disease, juvenile onset (ages 4-7)
- The disease is caused by a mutation in the CLN3 gene, found on chromosome 16. The gene directs the production of a protein called battenin, which is found in the membranes of the cell.
- Most children suffering from CLN3 disease have a missing part in the gene which in turn results in inability for the protein to be produced.
- Rapidly progressive vision loss begins between ages 4 and 7. Children develop learning and behavior problems, and slow cognitive decline (dementia) and then start having seizures around age 10. In the teenage years, children affected by CLN3 disease develop slow movement, stiffness, and loss of balance (also referred as parkinsonism).
- They also develop difficulty with speech and language.
- As they age, children and teenagers become increasingly dependent on their caregivers.
- Most children with the disease die between the ages of 15 and 30.
CLN4 disease, adult onset
- Also known as Kufs disease type B, this very rare form typically begins in early adulthood (normally around age 30) and causes problems with movement and early dementia.
- The symptoms progress slowly, and CLN4 disease does not cause blindness.
- It is related to mutations in the DNAJC5 gene on chromosome 20.
- The age of death varies among affected individuals.
CLN5 disease, variant late-infantile onset
- This disease is caused by problems with a lysosomal protein called CLN5, whose function is unknown.
- The CLN5 gene is located on chromosome 13. Children progress normally for the first few years of life before they start losing skills and develop behaviour problems.
- Seizures and myoclonic jerks begin usually between ages 6 and 13.
- Vision deteriorates and is eventually lost.
- Children have learning disabilities and problems with concentration and memory.
- Some may need a feeding tube.
- Most children with CLN5 live into their late childhood or teenage years.
CLN6, variant late-infantile onset
- The gene CLN6, located on chromosome 15, directs the production of the protein CLN6, also called linclin.
- The protein is found in the membranes of the cell (most predominantly in a structure called the endoplasmic reticulum).
- Its function has not been identified.
- Symptoms vary among children, but typically start after the first few years of life and include developmental delay, changes in behavior, and seizures.
- Children eventually lose skills for walking, playing, and speech.
- They also develop myoclonic jerks, problems sleeping, and vision loss.
- Most children with CNL6 die during late childhood or in their early teenage years.
CLN6, adult onset
- Also known as Kufs disease Type A, this form of CLN6 disease shows signs in early adulthood that include epilepsy, inability to control muscles in the arms and legs (resulting in a lack of balance or coordination, or problems with walking), and slow but progressive cognitive decline.
CLN7, variant late-infantile onset
- This disease is caused by mutations in the CLN7 gene located on chromosome 4, which produces the protein MFSD8—a member of a protein family called the major facilitator superfamily.
- This superfamily is involved with transporting substances across the cell membranes.
- As with all the other forms of Batten disease, the defect in the gene results in lack of production of the protein.
- Developmental delays begin after a few years of what seems to be a normally-developing child.
- Children usually develop epilepsy between the ages of 3 and 7, along with problems sleeping and myoclonic jerks.
- Children begin to lose the ability to walk, play, and speak as the disease progresses, with a rapid advancement of symptoms seen between the ages of 9 and 11.
- Most children with the disorder live until their late childhood or teenage years.
CLN8 disease with Epilepsy with Progressive Mental Retardation (EPMR)
- Abnormalities in the CLN8 gene cause epilepsy with progressive decline in mental function.
- The gene, located on chromosome 8, encodes a protein also called CLN8, which is found in the membranes of the cell—most predominantly in the endoplasmic reticulum (part of the recycling-bin machinery of the cell). The protein’s function has not been identified.
- Onset of symptoms begins between ages 5 and 10 and include seizures, cognitive decline, and behavioural changes.
- Seizures typically become very intermittent after adolescence. Loss of speech occurs in some individuals. Affected individuals can live into adulthood.
- A very rare form of the disorder is sometimes called Northern Epilepsy syndrome, because it occurs in certain families in an area of Finland.
CLN8 disease, late-variant onset
- Affected children begin showing symptoms between ages 2 and 7, which include loss of vision, cognitive problems, unsteadiness, myoclonic jerks, and behavioural changes.
- Children develop treatment-resistant epilepsy and a marked loss of cognitive skills by age 10.
- Many children lose the ability to walk or stand unassisted.
- Life expectancy is uncertain; some children have lived into their second decade of life.
CLN10 disease
- This very rare disease is caused by a mutation in the CTSD gene, located on chromosome 11, which produces a protein known as cathepsin D.
- Cathepsin D is an enzyme that breaks apart other proteins in the lysosome.
- The disease typically is seen soon after birth, although it can occur later in childhood or adulthood.
- Some children have microcephaly—an abnormally small head size with reduced brain size.
- In the congenital form, seizures may occur before birth but are hard to differentiate from normal baby movements.
- Following birth, babies may have seizures that do not respond to treatment, problems with breathing that can progress to respiratory failure, and obstructive sleep apnea.
- Babies may die shortly after birth or within the first weeks of life.
- A late-infantile form of the disease features a later onset of symptoms and slower disease progression.
- As children age, they develop seizures and progressive problems with vision, balance, and intellectual skills.Affected individuals also may have problems coordinating muscle movement and trouble with walking (called ataxia) as well as very stiff muscles (spasticity).
- Children with the disease often die in early childhood.
Diagnostic Tests
- Family medical history
- Neurological exam
- DNA analysis/genetic testing can confirm the presence of a mutated gene that causes an NCL disease, as well as be used in prenatal (before birth) diagnosis of the disease.
- Measurement of enzyme activity can be used to confirm or rule out CLN1 and CLN2 disease.
- Blood or urine tests.
- Skin or tissue sampling can show distinctive shapes formed by lipofuscin accumulation—some look like half-moons while others look like fingerprints—when viewed under a special microscope. The lipofuscins also take on a greenish-yellow colour when viewed under an ultraviolet light microscope.
- Electroencephalograms (EEG) monitor brain activity.
- Electrical studies of the eyes.
- Diagnostic imaging using computed tomography (CT) and magnetic resonance imaging (MRI) scans can help doctors look for changes in the brain’s appearance.